HUOM! Uusi osoite www.edssuomi.blogit.fi/luento , joka on virallinen sivustomme!
Dos. Ilkka Kaitilan luento Ehlers-Danlosin syndroomasta!
Ehelrs-Danlosin syndroomaa sairastaville tai sairaudesta kiinnostuville pidettiin viikonlopputapaaminen Reumaliiton kuntoutumiskeskuksessa Apilassa. Dos. Ilkka Kaitila perinnöllisyyslääketieteen lääkäri, HYKS:n yksiköstä luennoi Ehlers-Danlosin syndroomasta

(Luennon sai keskeyttää ja kysyä omia kysymyksiä kun halusi. Kaikkea ei ole kirjoitettu.)
Ehlers-Danlosin syndrooma (EDS) on harvinainen. Kun te tapaatte lääkärin, niin usein lääkärille nimi on kyllä tuttu mutta he eivät tiedä oikeastaan tästä sairaudesta mitään. Todellisuus on tietenkin se, että vuosikymmenten kuluessa on julkaistu tavaton määrä lääketieteellistä tutkimustietoa Ehlers-Danlos syndroomasta. Lääketieteellisessä tietokannassa vuoden 2008 oli 2467 julkaisua, joka luettelee nimenomaan tieteellisissä lehdissä olleet julkaisut. Suurin osa näistä julkaisuista on tällaisia potilastapauskuvauksia, joka on lääkäreille hyvin tärkeää, mutta niistä ei niin kuin potilaskohtaisesti opi useinkaan ns. yleissääntöjä joita voitaisiin käyttää kaikkien Ehlers-Danlos syndroomaa sairastavien hyväksi. Löytyy suhteellisen vähän sellaista lääketieteellistä tutkimustyötä, joka perustuu ns. suuremman joukon potilaiden tutkimuksiin. Se olisi varmasti hyvin tärkeää. Silloin löytyisi varsinaiset oikeat uudet linjat. Näitä tutkimuksia on vähän.
Kukaan ei tiedä kuinka monta Ehlers-Danlosin syndroomaa (EDS) sairastavaa potilasta Suomessa on. Meillä ei ole sellaista rekisteriä, jossa kaikki olisi lueteltu. Ehlers-Danlos monista muista sairauksista eroten on sillä tavalla ongelmallinen sairaus, että sen diagnoosin asettaminen ei ole ollenkaan helppoa. On aivan varmasti suuri joukko sellaisia henkilöitä tässä meidän maassamme, joilla ei ole diagnoosia Ehlers-Danlosin oireyhtymään, vaan heillä on jokin muu diagnoosi jos he ovat yleensä hakeutuneet terveydenhuollon selvityksiin. Useimmat Ehlers-Danlosin syndrooma sairastavat ovat sen tehneet, mutta heillä on erilaisia diagnooseja. Se johtuu tietysti siitä, että lääkärit eivät tunne Ehlers-Danlosin syndroomaa kovin hyvin, ei missään, ei Suomessa, ei missään maailmassa.
Kysymys: Onko näitä Ehlers-Danlosin syndroomaa sairastavia potilaita rekisteröity, joille on tehty varsinainen koepalamutaatiotutkimus geeneistä?
Mitään virallista rekisteriä heidänkään osaltaan ei ole. Riippuu siitä missä tuo tutkimus on tehty. Suomessa on Perinnöllisyyslääketieteen yksikköjä jokaisessa yliopistollisessa keskussairaalassa ja kaksi muuta näiden sairaaloiden ulkopuolella. Niissä usein pyritään tekemään varsinainen tarkka diagnoosi aina geenitutkimusta myöden, mutta Ehlers-Danlosin syndroomassa on se tilanne, ettei monissakaan muodoissa tiedetä mikä on se geeni. Ja nyt niilläkin potilailla, joilla tämä mutaatio on todettu, niin ei ole olemassa sellaista “velvoitetta” että eri yksiköistä lähetettäisiin tieto johonkin yhteen paikkaan. Eli nyt meillä on potilas, jolla on Ehlers-Danlos oireyhtymä tässä ja tässä geenissä on tämän ja tämän tapainen muutos. Tällaista velvoitetta ei ole. Jos koettaa selvittää Sosiaali- ja terveysministeriön tautirekisteristä - joka on kansainvälinen rekisteri – kuinka monta henkilöä on Suomessa, joilla on Ehlers-Danlosin syndrooma, niin ei pääse yhtään mihinkään. Ne ovat sielläkin aivan väärin rekisteröity. Monilla sairauksilla on tällainen potilasyhdistys ja usein siellä on se rekisteri. Ehlers-Danlos potilasyhdistystä ei ole vielä perustettu.
Reumaliiton kuntoutumiskeskus Apila Kangasalalla on Ehlers-Danlos oireyhtymää potevien keskus. On muutama muukin keskus esim. Reumasäätiön sairaala Heinolassa. Reumaliitto on julkaissut Ehlers-Danlos oireyhtymä –vihkosen jota on saatavilla Reumaliitosta.
Vihkosessa on kuitenkin yksi vika. Otsikossa sanotaan oireyhtymä, mutta itse asiassa kysymys on joukosta sairauksia jotka ovat aivan itsenäisiä sairauksia. Niissä on hieman samankaltaisia piirteitä ja siksi ne luetaan tähän samaan ryhmään, mutta kyse on kuitenkin erilaisista sairauksista.
- Niillä on tiettyjä yhteisiä piirteitä.
- Nivelten ylitaipuisuus
- Ihon ylivenyvyys
- Mustelma-alttius
- Kudosten repeilevyys (kudokset voivat repeillä eli liian pienestä vammasta johtuu se, että yhtäkkiä tuleekin haava tai kudokseen tulee halkeama)
- Haavojen huono paraneminen (haavat paranevat huonosti, hitaasti ja jättävät kummallisia arpia)
- Krooniset nivel-, raaja- ja lihaskivut
- Sydämen ongelmia, joista tavallisin on hiippaläpän prolapsi (prolapsilla tarkoitetaan sitä, että läppä antaa periksi ja verta ei mene ihan siihen suuntaan mihin pitäisi)
Nämä kaikki ovat ensinnäkin oireita, joita ei löydy suinkaan kaikilta. Nyrkkisääntönä melkein on, että ei löydy sellaista Ehlers-Danlos syndroomaa sairastavaa potilasta jolla olisi nämä kaikki oireet. Nämä ovat yleispiirteitä, eräänlaisia yhteisiä piirteitä. Ja sitten tulee sairaudesta riippuen näitä muita piirteitä. Kaikki nämä jo sinänsä viittaavat sidekudoksen heikkouteen jonkinnäköiseen semmoiseen, että kudos ei kestä sitä mitä sen pitäisi kestää.
Jos me pyrimme määrittämään mitä tarkoitetaan nivelten ylitaipuisuudella, niin sekään ei ole ihan selvä asia. Beighton on pyrkinyt määrittämään mitä tarkoitetaan ylitaipuisuudella tai ihmisellä jolla on ylitaipuisuutta eli hypermobiliteettia.
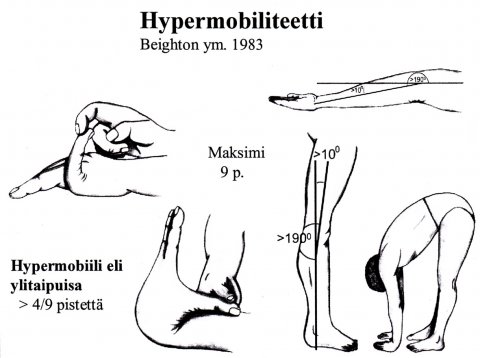
Kuvassa on viisi mitattavissa olevaa piirrettä, joille annetaan pisteet. Jos yhdeksästä piirteestä neljä on positiivista, niin silloin puhutaan ihmisestä jolla on hypermobiliteetti. Jo otamme joukon aivan terveitä varsinkin naisia tai nuoria tyttöjä niin aivan helposti he saavat 4 tai 5 pistettä. Tämä on myös ikäkysymys. On sellaisia aikuisia, joilla on lapsuudessaan ja nuoruudessaan ollut selkeästi hypermobiliteettia, mutta aikuisena ei enää ole. Näin tämä määritelmä miten hypermobiliteetti määritetään, niin se ole ihan yksinkertainen asia.
Ihon venyvyys. Kaikkien iho venyy kun otetaan ihosta kunnon ote ja vedetään tai nostetaan sitä. Ehlers-Danlos oireyhtymässä iholla on piirre, että se venyy liian paljon. Se riippuu paljon, mistä sitä koetetaan, mutta tuo standardipaikka on tuo kyynärvarsi ja kyynärvarren sisäsivu. On määritetty, että jos iho helposti venyy 2 cm, niin silloin se on ylivenyvä. Tähän liittyy vielä se että kun sitä venyttää, niin tulee sellainen tunne että siinä on sellaista kummallista kimmoisuutta, se antaa niin kuin kumimaisesti periksi ja sitten palaa takaisin. Vähän toisella tavalla, kuin terveen henkilön iho. Jos lääkäriltä jolla pitäisi olla sellainen koulutus, että he pystyvät tekemään diagnostista tutkimusta tutkien potilaita ja hän kokeilee onko potilaalla kimmoisa, venyvä, ylivenyvä iho niin se on vaikeaa. Se on hyvin subjektiivinen käsitys, että minkälainen ihon pitäisi olla.
On suuri joukko muitakin sidekudossairauksia, jotka pitää oikeastaan pois sulkea silloin kun koetetaan asettaa Ehlers-Danlos oireyhtymää. Tällaisia ovat esim. Marfan oireyhtymä, Osteogenesis imperfecta (synnynnäinen luustonhauraussairaus) mutta näilläkin potilailla hyvin usein iho on melko venyvä.
Kysymys: Onko mahdollista, että ihmisellä on sekä OI että Ehlers-Danlosin syndrooma?
Se on mahdollista. Mitä todennäköisimmin kysymys on, että hänellä on sellaisessa perintötekijässä sentyyppinen mutaatio, että se aiheuttaa sekä luumurtumat että venyvyyden.
Tunnetaan myös Cutis laxa, Epidermolysis bullosa, Pseudoxanthoma elasticum, Sticklerin oireyhtymä ja näitä on muitakin. Tämän listan kerron teille sen vuoksi, että silloin kun lääkäri joka uskoo tietävänsä näistä sairauksista jotain ja tekee diagnoosia, niin hänen täytyy pois sulkea nämä. Ja siinä on tietyt kriteerit ja hänen täytyy tuntea myös ne sairaudet.
Mitä on sidekudos? Emme usein tule ajatelleeksi, että itse asiassa siellä on satoja erilaisia valkuaisaineita, hiilihydraattirakenteita jotka muodostavat sellaisen verkon. Toisia aineita enemmän, toisia aineita vähemmän, osalla aineista on jokin tietty tehtävä, toisella taas toinen. Sidekudos muodostuu hyvin monista asioista, joista tärkein meidän kannaltamme on tuo kollageeni. Mutta sitten siellä on elastiinisäikeitä, joita niitäkin on useita erilaisia. On proteoglykaaneja, glykopreteiineja eikä tarvitse tarkalleen tietää mitä ne ovat muuta kuin että täytyy olla käsitys, että siellä hyvin paljon erilaisia aineita. Jos jossain valkuaisaineessa on esimerkiksi jokin rakenteellinen vika niin se muuttaa koko tämän sidekudoksen arkkitehtuuria siten, että siitä tulee yllättäviäkin oireita. Nyt kollageeni jonka sanoin että on tärkeä, niin itse asiassa näitäkin tunnetaan iso joukko. Niitä on ainakin 19 erilaista. Perusrakenne on sama, mutta niiden rakennetta määräävät erilaiset perintötekijät. Tiedämme, että tällä hetkellä on ainakin 29 perintötekijää, jotka määräävät aivan normaalien kollageenien rakennetta. On olemassa suuri joukko sellaisia mahdollisuuksia, missä virhe jossain aiheuttaa erilaisia ongelmia.
Kysymys: Onko sillä merkitystä, että saa kielenpään nenään kiinni?
Jos siellä on jokin virhe, niin ne oireet ovat juuri niitä yhteisiä Ehlers-Danlos –piirteitä kuten, venyvä-, kimmoisa-, veltto-, pehmeä iho, ihon repeilevyys, paperiarvet (tarkoitetaan sellaisia ihan ohuita arpia), nivelten ylitaipuisuus tai taipumus nivelten sijoiltaanmenoihin, verisuonirepeämiä, suoli- ja rakkorepeämiä tai divertikkeleitä (divertikkelit ovat kuin pusseja, jotka tulevat putkimaisen tai rakkomaisen rakenteen sivuun), suonikohjut, luumurtumat ja niitä on monenlaisia. Kun tällaisia piirteitä näkee niin kuin tällaisena terveydellisenä harmina, niin voi epäillä että on kysymys sidekudoksen jonkun rakenteen virheestä.
Kysymys: Missä näitä luumurtumia voi olla, jos kysymys on sidekudoksen heikkoudesta?
Osteogenesis imperfecta on hyvä esimerkki.
Kysymys: Voiko se selkään tulla murtumia?
Voi. Hyvinkin. Nikamien kokoonpainumia.
Luu muodostuu tietysti tällaisesta pehmeästä orgaanisesta aineesta, jossa on runsaasti tietynlaista kollageenia ja paljon muita aineita. Kovaksi sen luun tekee mineraalit hiili kaiken kaikkiaan, kalsiumkarbonaatti ja monet muut mineraalit. Jos kollageenissa eli tässä perusaineessa on virhe, niin silloin se että luu on kovaa niin se ei auta, murtuma-alttius siitä tulee. Luu ei kestä sellaista rasitusta mitä sen pitäisi. Ehlers-Danlos oireyhtymissä murtumat eivät ole pääongelma, mutta Osteogenesis imperfectassa on.
Monenlaista voi odottaa, jos sidekudoksessa on virhettä. Sidekudoksen heikkousoireet voivat olla hyvin kummallisia. Esim. jollain voi olla, että silmäluomet voi “kääntää nurin” hyvin helposti. Kaikilla ihmisillä kyllä tämä on mahdollista, kun on ihminen joka osaa sen tehdä eli sellaisella puikolla käännetään.
On. Robert J. Gorlin (1923 - 2006) amerikkalainen, alun perin hammaslääkäri ja hänestä kehittyi aivan tavattoman hyvä perinnöllisten sairauksien tuntija. Tämä oire, että kieli ulottuu nenän päähän, niin sitä kutsutaan Gorlinin ominaisuudeksi. Se on yksi näitä Ehlers-Danlos oireyhtymän piirteitä, mutta on pitkäkielisiä ihmisiä joilla ei ole Ehlers-Danlosia vaikka kieli ulottuisikin nenään.
Kysymys: Voiko skolioosi muuttua ajan myötä, muuttua toiseen suuntaan?
Useimmin on niin, että ruumin rakenne pyrkii korjaamaan tällaisen virheasennon ja selkärangassa nimenomaan eli jos selkäranka lähtee menemään yhteen suuntaan, niin se kompensatorisesti tekee vähän ylempänä toiseen suuntaan sen käyrän. Skolioosi on yksi Ehlers-Danlos oireyhtymän ongelmista, ei onneksi kovin hankala. On monia sairauksia, joissa se on paljon vaikeampi. Lapsuusiällä sitä ei tarvitse olla, vaan se lapsuus- ja kasvuiässä sitten alkaa kehittyä. Kun kasvuikä on päättynyt, niin silloin skolioosi muuttuu enää vähän. Se voi muuttua jonkin verran, mutta aika vähän. Se on nimenomaan kasvuiässä ilmenevä.
Kysymys: Jos välilevy katkeaa tai repeää ilman mitään tapaturmaa?
Tarkoitat varmasti sitä, että välilevystä purskahtaa vähän se kiinteämpi rustoinen aineosa sieltä ulos eli se on repeäminen, prolapsi. Selkärangan välilevyillä on oma kestävyytensä elämän aikana ja vaikka varsinaista sellaista kovempaa tapaturmaa ei tarvitse tulla, niin onhan selkä koko ajan tietyn rasituksen alaisena. Kun ikää tulee, niin voi käydä näin.
Kysymys: Voiko Ehlers-Danlosin syndroomaa sairastavalla potilaalla tulla tällaisia vammoja, niin liian rajun kuntoutuksen perusteella.
Kyllä sellainenkin mahdollisuus on. Fysiatrit, kuntoutuslääkärit ja lääkintäjumpparit tuntevat Ehlers-Danlos oireyhtymän yleisesti aika tavalla huonosti ja voivat koettaa noudattaa niitä tavallisia kuntoutuksen periaatteita, jotka eivät ole oikeita Ehlers-Danlos oireyhtymää sairastaville. Hyvä esimerkki on, että venyttely ei ole kovinkaan hyvä asia Ehlers-Danlos oireyhtymää sairastavalle. Kun sitä vähän ajattelee, niin ei se ole mikään ihme, mutta ei sitä tulla ajatelleeksi edes kuntoutuslaitoksissa.
Kommentti kuulijalta: Lääkäri totesi, ettei hänelle sovi kuntoutus. Kuntoutus oli aiheuttanut hänelle selkärankaan vahinkoa.
Juuri tällainen mahdollisuus on. Sen takia kun harvinaisesta sairaudesta on kysymys, niin se mitä te itse tiedätte ja mitä te välitätte siitä lääkäreille, hoitohenkilökunnalle, kuntouttajille on erittäin tärkeää.
Historiaa
Ehlers-Danlos oireyhtymä ei ole mikään äskettäin todettu sairaus, vaan kuvauksia löytyy jopa vuosituhansien takaa. Silloin ne on kuvattu patsaan, maalauksen tai piirroksen muodossa. 1600-luvulla oli ensimmäinen lääketieteellisessä julkaisussa kuvaus. Tschernogobow (1892) mainitaan aina ensimmäisenä, joka varsinaisesti kuvasi Ehlers-Danlos oireyhtymän. Hän ei kuitenkaan tälle oireyhtymälle saanut tuota nimeään. Tunnetaan paljon harvinaisia sairauksia. Kun joku lääkäri toteaa tai tutkija toteaa sairauden ja julkaisee sen ja se huomataan, niin siitä tulee hänen nimellään kulkeva oireyhtymä. Näin on juuri Ehlers-Danlos oireyhtymässä käynyt. Tanskalainen ihotautilääkäri edellisen vuosisadan alussa Edward Ehlers (1901) kuvasi ensimmäisen potilaan. Joitakin vuosia myöhemmin ranskalainen ihotautilääkäri Henri-Alexandre Danlos (1908) kuvasi toisen ja tästä tulee Ehlers-Danlos oireyhtymä.
Monet Ehlers-Danlos oireyhtymän piirteet ovat epätavallisia, kummallisia, ihmeellisiä, niin monet potilaat ovat käyttäneet sitä hyväkseen. He eivät ole ehkä jossain tavallisessa fyysisessä ammatissa toimia, niin he ovatkin lähteneet esiintymään. Tunnetaan “The Indian Rubber Man” eli Kumimies, “The Elastic Lady, “The Human Pretzel” kertoo myös tällaisesta ylitaipuisuudesta. He ovat tehneet elantonsa tietyn ajanjakson jossain sirkuksessa ja saanet sillä tavalla elantonsa.
Yleisyys
Kuten alussa kerroin, niin ei ole rekistereitä. Ei Suomessa, ei missään. Suomen terveydenhuolto on paljon paremmassa tilassa kuin monissa muissa maissa, koska meitä täällä on vähän ja terveydenhuolto on järjestetty hyvin ja pyritään selvittämään mitä sairauksia Suomessa. Muualla on paljon huonompi tilanne.
Uskotaan, että Ehlers-Danlos oireyhtymää löytyy 1: 5 000 – 1: 10 000. 1 : 10 000 merkitsee noin 500 hlö Suomessa. Se on vähän. Toistaiseksi ei vielä ole Ehlers-Danlosin yhdistystä. Minun tiedossani on 50 - 60 henkilöä joilla on Ehlers-Danlos oireyhtymä. Se on 1/10 mitä täällä Suomessa todennäköisesti on.
EDS - Yleisyys
- Harvinaisia – tavallisia, tiedot, arvioita
- 1:5 000 – 1:10 000 (kaikki tyypit)
- EDS III tavallisin ?
- Ei väestöeroja ?
- Ei sukupuolieroja ?
- Eliniän odote: lähes normaali
- Vaskulaari EDS IV – keskim. elinikä n. 50 vuotta
Ehlers-Danlosin oireyhtymän luokittelu
Ehlers-Danlosin syndroomassa on useita tyyppejä. Mikä niistä on tavallisin se on epäselvää. Eräät tutkimukset joissa on tietyltä ajanjaksolta yhdestä yksiköstä analysoitu minkä tyyppisiä potilaita heillä on ollut niin päätyvät siihen, että ylitaipuisuustyyppiä eli Ehlers-Danlos tyyppiä III olisi eniten. Toinen tutkimuskeskus jossa päädytään siihen, että ns. klassista muotoa on eniten. Kukaan ei tiedä mikä on se totuus.
Toiset kirjoittajat ovat sitä mieltä että väestöissä ei ole mitään eroa. Ehlers-Danlos syndroomaa on kaikissa väestöissä kun alettaisiin tutkia tarkemmin. Epäselvä asia. Todennäköisesti totta, että väestöissä ei ole suurempaa eroa.
Onko sitten sukupuolieroa. Kun ajatellaan potilaita, niin miehet hakeutuvat harvemmin tutkimuksiin ja hoitoon. Onko niin että sitä olisi miehillä lievempänä vai mikä siinä on, mutta näyttää kuitenkin siltä että Ehlers-Danlos on enemmän kyllä naisten ongelma. Sillä on itse asiassa juuri se tausta, että tytöt ja naiset ovat kudoksiltaan pehmeämpiä kuin miehet. Naiset myös hakeutuvat helpommin hoitoon.
Tärkeä kysymys on tietysti se, että vaikuttaako tämä elämänkulkuun. Yksi mitta on tietysti eliniän pituus. On sellainen käsitys, että nämä tavallisemmat Ehlers-Danlos oireyhtymän tyypit eivät oleellisesti siihen vaikuta. Ehlers-Danlos tyyppi IV eli vaskulaarinen muoto, niin siinä selvästi ja aiemmin vielä selvemmin elinikä oli lyhentynyt. Johtuu siitä, että siihen voi liittyä juuri sentyyppisiä komplikaatioita että se johtaa kuolemaan ennenaikaisesti.
Erityisesti perinnöllisyyslääkärit , mutta jonkin verran myös ortopedit ovat pyrkineet luokittelemaan Ehlers-Danlos oireyhtymät ja useitakin tällaisia luokitusyrityksiä on tehty, se viimeisin luokittelu on noin kymmenen vuoden takaa. Se perustuu tietysti sairauspiirteisiin, mutta myös periytymistapaan ja perussyyhyn kun se on saatu selvitettyä, näihin kolmeen perusasiaan. Käytännössä diagnoosin asettaminen ei ole kovin vaikeaa kun tiedetään mistä on kysymys. Silloin kun iho on ylivenyvä, niin ei pitäisi olla vaikeaa. Mutta sitten on paljon lievempiä ongelmia, terveydellisiä harmeja. Sitten on tämä ikäkysymys, lapsuusiässä diagnoosin asettaminen on paljon vaikeampaa. Silloin diagnoosin asettaminen ei olekaan enää ihan helppoa. Tosiasia myöskin on, että vaikka olisi hyvin paneutunut tuohon Ehlers-Danlos oireyhtymään, niin sen tarkan, luotettavan luokituksen asettaminen yhdelle potilaalle on vaikeaa. Monet julkaisut joissa yhteenvedonomaisesti Ehlers-Danlos oireyhtymästä kerrotaan, niin päätyvät että vain noin puolessa se onnistuu.
Kysymys: Olen kuullut että vaikka monelle olisikin tehty biopsia, niin siitä huolimatta he eivät mahdu ns. johonkin tiettyyn tyyppiin. Voiko EDS varioida niin paljon, että esim. II tyypin Ehlers-Danlosin syndroomassa saattaa olla myös III tyypin oireita? Diagnoosini on EDS II (mahdollisesti III).
Kyllä. Nimenomaan se voi varioida niin paljon. Nämä oireet menevät monta kertaa niin päällekkäin.
Kysymys jtk: Eli kyse ei voi olla siitä osaako lääkäri asiaansa vaan tilanteesta ei voi ottaa selvää.
Juuri näin. Asiantuntijallekin vain noin puolessa tapauksissa tyypin luokitus aivan varmasti onnistuu. Jos meillä olisi joku laboratoriotesti kuten esim. geenitesti, niin silloin se onnistuisi useimmin. Nyt monessa muodossa ei edes tiedetä mitä geeniä katsottaisiin. Mistä etsittäisiin sitä mutaatiota, ihmisellä on 25 000 geeniä.
Kysymys: Minulta on 80-luvun alkupuolella tehty koepalatutkimus, jossa todettu että kollageeni on ollut viallinen. Onko mahdollista, että kun on kaikki oireet sidekudoksessa niin että sitä ei olisi luissa?
On. Kyllä se on mahdollista. Johtuu siitä, että kollageeneja on 19 erilaista. Ne ovat kudoksesta riippuen jakautuneet. Luukudoksen ja ihon kollageeni vaikka ne ovat samankaltaisia, niin ne ovat kuitenkin jonkin verran erilaisia. Ihosta näytteen ottaminen ei tuo diagnoosia. Siis en voi sanoa, että kollageenissa on vikaa ja kysymyksessä on todennäköisesti Ehlers-Danlos oireyhtymästä, mutta se ei sano luokittelusta mitään.
Kysymys: Minulla on Ehlers-Danlos III – IV. Sukutausta viittaisi neljänteen, koska suvussa on aortan repeämää. Onko sitä mahdollista tutkia?
Varmasti olisi syytä mennä. Olisi syytä mennä perinnöllisyyslääketieteen yksikköön ja pyrkiä sitä selvittämään.
Kysymys: Eli sekin, että jos on EDS III tai IV niin sitä ei välttämättä pystytä varmistamaan millään testeillä.
EDS IV:n tällä hetkellä pystyy erottamaan geenitestillä, mutta EDS tyyppi III:ssa ei tiedetä mikä on se perintötekijä. On pieniä aavistuksia mikä se voisi olla mutta ei ole täyttä varmuutta. Olisi tärkeää myös tämän periytymisen kannalta tietää että mikä muoto on kyseessä. Silloin siihen tulee sukulaiset. Jos ei ole selvää että tuleeko se omalta vanhemmalta, niin silloin tulee kysymys entäs minun jälkeläiset.
Kysymys: Minulla on todettu EDS kliinisillä oireilla ja niillä tiedoilla mitä äidilläni on. Onko tilanne se, että jos äidilläni on Ehlers-Danlos II, niin silloin minullakin on oltava EDS II?
Kyllä.
Kysymys jtk: Eli siis tyyppi ei voi vaihtua?
Tyyppi ei vaihdu, mutta käsitys siitä mikä tyyppi se on loppujen lopuksi, niin voi olla että se ei ole niin varma.
Kysymys jtk: Voiko se varioida?
Kyllä. Se voi varioida. Äidilläsi voi olla hieman erilaiset oireet kuin sinulla. Yhtenä syynä on yksinkertaisesti ikäero.
Kysymys: Voiko vasta vanhemmiten tulla jotain oiretta esim. sydänoire, jota tähän mennessä ei ole ollut?
Voi tulla. Tämä on myös asia, joka pitää ymmärtää.
Kysymys jtk: Voidaanko sitä tutkia etukäteen?
Tiettyjä asioita pitääkin tehdä, tutkia ja seurata ja tietyllä tavalla pois sulkea komplikaatio-ongelmien kehittyminen. Se on tärkeää. Ehlers-Danlos oireyhtymää sairastavalle ei riitä se, että hänet kerran tutkitaan. Hänellä pitää olla jonkinlainen seurantasysteemi, jossa tiettyjä asioita seurataan. Katsotaan vuoden kuluttua, kahden vuoden tai viiden vuoden riippuen siitä, mistä ongelmasta on kysymys. Tämänkin takia juuri tällaiset keskukset ovat tärkeitä.
Niin teistäkin kuin meistä lääkäreistä jotka Ehlers-Danlos oireyhtymää pyrimme hoitamaan, niin olisi tärkeää, että luokittelu olisi varma. Voisi sanoa, että sinulla on tämä. Tästä on kysymys. Käytännössä sillä ei itse asiassa ole suurta merkitystä, sillä saman luokankin sisällä oireet vaihtelevat, ennuste vaihtelee, se mitä pitää tehdä ja ei pidä tehdä vaihtelee. Tärkeintä olisi juuri että olisi keskuksia, joissa olisi sellaisia lääkäreitä, lääkintäjumppareita, sairaanhoitajia ja sosiaalihoitajia joilla on paljon kokemusta Ehlers-Danlos oireyhtymästä, jolloin he voivat sitä potilasta tutkiessaan päätellä mikä on tälle potilaalle parasta.
EDS – Luokittelu
· Viimeisin luokittelu 1997
o perustuu sairauspiirteisiin, periytymistapaan ja perussyyhyn
· Käytännössä EDS diagnoosin asettaminen ei ole vaikeaa – jos tuntee oireyhtymän
· Luokittelu onnistuu asiantuntijaltakin vain n. 50%:a tapauksista
Luokittelu
Diagnostiset kriteerit käyn läpi lyhyesti että nähdään, minkälaisia Ehlers-Danlos oireyhtymän tyypit ovat. Mitkä ovat pääkriteerit, mitkä sivukriteerit. Tästä ehkä rupeaa hahmottumaan, minkälainen sairaus teillä itsellänne on. Alla luettelen pääkriteerit ja sivukriteerit. Sivukriteerit eivät ole niin tärkeitä tämän diagnoosin kannalta.
Kysymys: Montako sivukriteeriä pitää täyttyä Klassisen tyypin diagnoosiin?
Ne on kaikkien näiden muotojen mukaan määritetty jonkinnäköisellä tarkkuudella. Pitäisi olla 2 - 3 pääkriteeriä ja ehkä 1 – 2 sivukriteereitä. Kun katsotaan minkälaisia nämä muiden sairauksien pääkriteerit ja sivukriteerit ovat, niin huomataan se että miten määritetään, niin on hyvin pitkälti subjektiivinen asia.
EDS-Villefrance 1997 –luokittelu 10 EDS tyyppiä
· Klassinen EDS I ja EDS II
· Hypermobiili EDS III
· Vaskulaarinen EDS IV
· Kyfoskolioottinen EDS VI
· Artrokalaattinen EDS VIIA ja EDS VIIB
· Dermatosparaktinen EDS VIIC
· Muut EDS V, EDS X, EDS tenaskiini-X

Klassinen Ehlers-Danlos oireyhtymä I ja II
Dg -pääkriteerit:
· Ihon ylivenyvyys
· Haavojen huono parantuminen
· Ihon laajat, atrofiset arvet
· Nivelten ylitaipuisuus
Dg -sivukriteerit:
· Pehmeän samettimainen iho
· Mustelma-alttius
· Molluskatuumorit, ihonalaiset “herneet” (ihon alla olevat pyöreähköt pehmeät kyhmyt, joita kutsutaan molluskoiksi, eräänlaiset “herneet”)
· Lihasheikkous ja lihasvoiman heikkous (hypotonia)
· Nivelluksaatiot, lattajalka
· Leikkausten jälkiongelmat erityisesti kudosrepeämät
· Tyrät, prolapsit, keskenmenot
· Sukutiedot (vallitseva periytyminen)
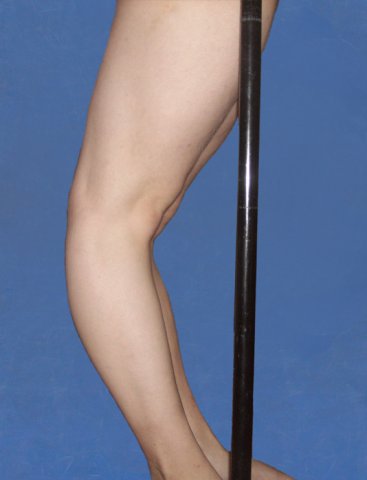
Atrofia merkitsee sitä että arpi muodostuu kummallisen ohueksi. Arvet voivat olla tavallista suurempia ja laajoja. Varsinkin pojille tulee arpia ja haavoja otsan seutuun pienestä kaatumisesta. Iho venyy (kuva hieman alempana). Pituuteen tämä muoto ei vaikuta. Se ei aiheuta luuston deformiteetteja. Klassisessa tyypissä esim. pieni lapsi voi saada polvensa taipumaan aivan merkillisellä tavalla. Synnynnäinen lonkkaluksaatio saattaa myös tulla. Polviin ja alaraajoihin saattaa tulla paperimaisia, ohuita, pehmeitä arpia. Säärissä olevat ovat usein seurausta siitä, että pienistä vammoista ihon alle syntyy aika laajakin verenvuoto ja se imeytyy hitaasti pois, mutta jättää sitten ihan ohuet ihoarvet.
Nämä on tyypillistä juuri Ehlers-Danlos tyyppi I:lle ja vähän lievempänä EDS tyyppi II. Ientulehdus on aika harvinainen ja hankala ongelma. Ikenet saattavat tulehtua hyvinkin pahasti ja ne ovat vaurioittaneet myös hampaistoa. EDS tyyppi I:ssä ja periodontaali-muodossa tämä on tyypillinen piirre.
Luokittelun tekeminen potilaskohtaisesti ei ole helppoa. Tyyppi I – II muodostavat ns. klassisen Ehlers-Danlos oireyhtymän. Tästä muodosta löytyy juuri tällaisia vanhoja kuvauksia kuten “The Rubber Man” sairasti jompaakumpaa näistä tyypeistä. Näiden ero on, että tyyppi I on vaikeampi ja II on lievempi, mutta ne aiheutuvat saman perintötekijän virheestä. COL5A1 ja COL5A2 mutaatioita (37,2 %)

Hypermobiili Ehlers-Danlos III
Dg -pääkriteerit:
· Nivelten ylitaipuisuus
· Pehmeä, samettimainen iho
Dg -sivukriteerit:
· Toistuvat nivelluksaatiot
· Hankalat, vaihtelevat, krooniset nivel- ja raajakivut,(jotka paikantuvat niveliin, selkään, lihaksistoon)
· Sukutiedot (vallitseva periytymistapa)
Ehlers-Danlos oireyhtymän tyyppi III on nivelten ylitaipuisuusmuoto. Tämä Hypermobiilioireyhtymä tai Ehlers-Danlos III menevät aika pahasti päällekkäin sairausryhmän kanssa, jota kutsutaan juuri Ylitaipuisuusoireyhtymäksi. Lääkärit ja tutkijat ovat hieman epätietoisia siitä, että onko kysymys ihan samasta asiasta. Dos. Ilkka Kaitila ajattelee juuri näin. Itse asiassa tämä Hypermobiilioireyhtymä joka on ihan tavallinen ongelma, on sama asia mutta lievempi kuin Ehlers-Danlos oireyhtymä tyyppi III. COL3A1 (?) mutaatioita.
Kysymys: Onko sillä kuitenkin eri nimi?
Nykyisin jo laitetaan yhtäläisyysviivaa siihen väliin.
Kommentti: Ymmärtääkseni Britanniassa on tämä sama linja. Jos Hypermobiliteettisyndrooma on “pahan oloinen”, niin silloin yleensä annetaan Ehlers-Danlos diagnoosi.
Kyllä.
Olympiakisojen voimistelussa näkee huippu-urheilijoita tavallisesti pikkutyttöjä, jotka taipuvat uskomattomiin asentoihin. Tuskin heillä on Ehlers-Danlos oireyhtymää.
Vaskulaari Ehlers-Danlos IV
Dg -pääkriteerit:
· Ohut, läpikuultava iho (iho melko kovaa, venyminen ei ole sinänsä piirre)
· Valtimo-, suolisto- ja kohturepeämät
· Vaikea mustelma-alttius
· Tyypilliset kasvojen piirteet
Dg -sivukriteerit:
· Sormien ja varpaiden suippous (acrogeria)
· Pikkunivelten ylitaipuisuus
· Jänne- ja lihasrepeämät
· Kampurat
· Laskimolaajentumat
· Arterio-venoosit fistulat (valtimoiden ja laskimoiden väliset suorat yhteydet joita kutsutaan fistuloiksi)
· Ilmarinta
· Ienvetäymät
· Leikkausten jälkiongelmat
· Sukutiedot, erityisesti läheisten äkkikuolemat
· Periytyy vallitsevasti
Tämä Vaskulaarinen eli verisuoni Ehlers-Danlos oireyhtymä on sillä tavalla kaikkein tärkein, että tämä on hengenvaarallinen sairaus. Siinä iho on ohutta, läpikuultavaa, itse asiassa aika kovaa. Siinä tämä venyminen ei ole piirre. Valtimoiden, suoliston ja kohturepeämät saattava olla aivan yhtäkkiset silloin kun niihin joku rasitus kohdistuu. Tavallisesti kyse on suurista valtimoista. Se voi olla aortta tai aivoverisuoni. Arvaatte, että ne ovat hengenvaarallisia tilanteita. Tyypilliset kasvon piirteet löytyvät erilaisista kirjoista. Pitkä kapea nenä, (mutta se on vähän epäselvä, sitä ei ole kaikilla). Kerron henkilöistä, jolla on Ehlers-Danlos tyyppi IV. Voin kertoa joistain seuraavia piirteitä. Yhdellä oli huonot, hennot, huonosti kehittyneet hiukset, jota ei tähän EDS oireyhtymään muutoin lueta, muuta hän joutui käyttämään peruukkia. Hänen äitinsä kuoli 42-vuoden iässä äkillisesti. Kuolinsyyselvityksessä osoittautui että hänen aorttansa oli revennyt. Joillain on ollut siinä 40 vuoden iässä aivoverisuonirepeämä, joka saatiin hoidettua eikä siitä jäänyt mitään halvaus- tai muita oireita. On tapahtunut suoliston repeämisiä. EDS tyyppi IV sormet ja varpaat ovat yleensä suipot ja laskimoiden pitäisi näkyvä varsin hyvin ainakin jaloissa. Ne kuultavat ohuen, vähän kovan ihon läpi. Varpaat ovat jollain tavalla resorpoituneet, hävinneet, sulaneet osittain pois, joka on myös tämän oireyhtymän piirteitä. Pikkusormikin saattaa olla hyvin pieni, sulanut pois.
COL3A1 mutaatioita.
Kysymys: Isälleni tuli aortan repeämä 74-vuotiaana, niin onko vain huonoa tuuria vai voisiko olla Ehlers-Danlos tyyppi IV? Hän oli HYKS:ssa sydänpoliklinikalla. Koska olen itse ollut tutkimuksissa Perinnöllisyysklinikalla, niin kulkeeko tästä isäni tapauksesta tieto Perinnöllisyysklinikalle?
En ole selvillä miten se tieto kulkee, mutta aortan repeämä voi johtua hyvin monesta syystä. Yksi tavallisin on kuitenkin Marfan oireyhtymä. Marfan oireyhtymässä aortta repeää sisäkalvoltaan ja tavallisesti se nouseva aortta, joka lähtee sydämestä niin kuin nousemaan ensin ja sitten laskee alaspäin. On toinen muoto, jossa repeää vatsa-aortan sisäkalvo ja se on myös perinnöllinen, mutta se ei ole Ehlers-Danlos oireyhtymä kuitenkaan. Nyt on mahdoton tietysti sanoa, että onko hänellä, onko sinulla Ehlers-Danlos oireyhtymä.
Kommentti jtk.: Minulla on diagnosoitu Ehlers-Danlos oireyhtymä. Onko se sitten tyyppiä III vai IV, niin sitä ei tiedetä. Pitäisikö sitä tyyppiä selvittää?
Ehlers-Danlos tyyppi IV täytyy ilman muuta pyrkiä selvittämään. Se että isäsi näin myöhäisellä iällä saa aortan repeämän viittaa siihen, että ei olisi kysymys tyyppi IV. Se on tavallisesti hieman nuoremman iän ongelma.
Tässä EDS tyyppi IV se vika on tyypin III kollageenin geenissä. Näitä kollageeni geenejä tunnetaan kaksi. Tässä toisessa, joka lyhennetään COL3A1, niin tässä toisessa tämän ihmisen geeneistä (kun niitä kaikkia geenejä on siis kaksi kappaletta) on mutaatio. Tämä kollageeni muodostuu kolmesta ketjusta, jotka kiertyvät perusmolekyyliksi ja näitä molekyylejä sitten pinoutuu ja muodostuu kudosta jota on juuri verisuonten seinämissä. Kun kollageenin geeni alkaa solussa/sidekudoksessa toimia, niin molemmat näistä perintötekijöistä tuottavat tällaista ketjua ja nämä kiertyvät sitten yhteen. Ne muodostavat eräällä tavalla sattumanvaraisesti sitä kollageenia sillä tavalla, että 1/8 siitä muodostuvasta kollageenista muodostuu vain siitä virheellisistä ketjuista. 3/8 muodostuu sellaisesta, jossa vain toisessa on se virhe ja 3/8 sellaisesta, jossa pienempi osa on tuota virheellistä. Vain 1/8 tuosta muodostuvasta kollageenista on normaalia. Tämä epäsuhta, että siellä on niin paljon tätä virheellistä kollageenia aiheuttaa tuon vaskulaarisen Ehlers-Danlos tyyppi IV.
Tällä hetkellä voidaan tehdä se tutkimus eli etsitään onko tuossa perintötekijässä virhe. Se on mutaatiotutkimus, jota Suomessa ei kukaan tee. Näyte lähetetään ulkomaille. Tänä päivänä se ei ole ongelma. Se voidaan lähettää sinne kun epäillään, että tässä nyt vikaa olisi. Kallis tutkimus, mutta ilman muuta se pitää ja kannattaa tehdä silloin, kun tätä tautia epäillään.
Kyfoskolioottinen Ehlers-Danlos VI
Dg -pääkriteerit:
· Nivelten ylitaipuisuus
· Vaikea lihasheikkous (hypotonia) jo vastasyntyneenä
· Etenevä selkärangan virheasento (progressiivinen skolioosi)
· Sarveiskalvon, silmämunan puhkeaminen
Dg -sivukriteerit:
· Kudosrepeämät
· Mustelma-alttius
· Valtimorepeämät
· Osteopenia, jolla tarkoitetaan luun mineraalikato
· Hoikka ruumiin rakenne
· Sukutiedot (peittyvä periytymistapa)
Kyfoskolioottinen Ehlers-Danlos tyyppi VI eli selkään paikantuva. Tässä pääongelma on selkärangassa. Skolioosi eli selän virheasento, käyryys. Kyfoosi tarkoittaa kumaraharteisuutta. Tässä on erityisen suuri riski myös sarveiskalvon ja silmämunan repeämiseen jo pienistäkin vammoista. Aikaisemmin on puhuttu myös okulaarisesta eli silmään paikantuvasta kyfoskolioottisesta tyypistä VI. Tämä periytyisi peittyvästi eli vanhemmat ovat terveitä, mutta lapsilla on 25% riski saada tätä sairaus.
Tätä kyfoskolioottista tyyppiä voisi kuvata seuraavilla sanoilla hyvin hoikka, kumaraharteinen. Vahvasti likinäköinen. Skolioosin ei tarvitse olla kovin paha, mutta selkäranka voi olla täysin poikkeava. Paperiarvet ovat hyvin tyypillisiä vammojen seurauksena.
Lysyloksidaasin puutos, PLOD1 mutaatioita.
Artorokalaattinen Ehlers-Danlos VIIA ja VIIB
Dg -pääkriteerit:
· Synnynnäinen lonkkaluksaatio
· Vaikea nivelten ylitaipuisuus
· Toistuvat nivelluksaatiot
Dg -sivukriteerit:
· Ihon ylivenyvyys
· Kudosten repeilevyys
· Mustelma-alttius
· Kyfoskolioosi
· Osteopenia
· Sukutiedot (peittyvä periytymistapa)
Artrokalaattinen muoto eli nivelten sairaus. EDS VIIA ja VIIB viittaa nivelten erityisiin ongelmiin. Synnynnäinen lonkkaluksaatio on hyvin tavallinen. Yleisesti nivelten ylitaipuisuus on vaikeaa, joiden seurauksena nivelten paikoiltaan meno eli luksaatio.
EDS tyyppi jaetaan kahteen erilaiseen muotoon. COL1A1 ja COL1A2 mutaatioita.
Dermatosparaktinen Ehlers-Danlos VIIC
Dg -pääkriteerit:
· Vaikea ihon repeilyvyys
· “Runsas” iho
Dg -sivukriteerit:
· Pehmeä iho
· Mustelma-alttius
· Napa- ja nivustyrä
· Sukutiedot (peittyvä periytymistapa)
Dermatosparaktinen EDS VIIC, jossa pääongelmat ovat ihossa. Ihon repeilyvyys on hyvin vaikeaa. Kädet eroavat oleellisesti EDS tyyppi IV –muodon käsistä. Sormet ovat lyhyet, mutta ihoa on liikaa. Jaloissa on tyypillisiä ohuita arpia ja iho on kaiken kaikkiaan velttoa.
Tässä tyyppi on VIIC. Prokollageeni N-proteinaasin puutos, ADAMTS2 mutaatioita.
Sitten on vielä joukko muita tyyppejä.
EDS tyyppi V
Tyyppi V oli olemassa mutta poistettiin, koska todettiin että se ei itse asiassa oireiltaan sovi Ehlers-Danlos oireyhtymäksi. Se luokitellaan epävarmaksi senkin takia, että näitä potilaita on ehkä kuvattu yksi. Yksi potilas, joka ei oikein sovi tähän mihinkään niin se on silloin tyyppi V.
EDS X – Tenaskiini-X:n puutos, jossa X ei merkitse nro kymmentä.
EDS – sydän- ja verisuoniongelmat
· Riippuvat EDS – tyypistä ja aineistosta – suuret vaihtelut
· Hiippaläpän prolapsi 6 – 78 %
· Läppävuoto 26 %
· Laajentunut aortan tyvi 13 – 33 %
· Poikkeava sydänfilmi 63
· EDS IV – suurten valtimoiden repeämisvaara
· EDS – potilailla tulee olla säännöllinen seuranta (UÄ, EKG, MRI) 1 – 5 vuoden välein Huom!
Tyyppi IV on ihan oma sairautensa riskinsä suhteen. Myös Klassiseen muotoon kuuluu esim. hiippaläpän prolapoitumista ja erilaisia sydämen läpän muotoja. Sydämessä on neljä läppää. Hiippaläppä (mitraaliläppä), eteisten väliset läpät ja keuhkovaltimoläppä. Erilaiset tutkimukset kertovat erilaisista esiintymisistä näissä läppäongelmissa. Ne vaihtelevat paljon tutkimusaineistosta riippuen 6 – 78 %. Nämä prosentit on mitä epäluotettavimpia sen takia, että ei ole luotettavasti tutkittuja perusaineistoja josta lähdettäisiin tätä selvittämään.
Läppävuoto on ominaisuus, joka kehittyy iän myötä. Sitä on 26 % yhden tutkimuksen mukaan. Laajentunut aortan tyvi joka on riski sillä, että sen sisäkalvo repeää. Tässäkin esiintyminen vaihtelee 13 – 33 %. Jos tehdään tavallinen sydänfilmitutkimus, niin yli puolella Ehlers-Danlos potilaista on sydänfilmissä jonkinnäköisiä poikkeavuuksia.
Ehlers-Danlos potilaan seuranta:
Suurten valtimoiden repeämisvaara erityisesti tässä EDS tyypissä IV, mutta se on olemassa myös näissä muissa muodoissa vähäisempänä. Loppuun olen laittanut ohjeen, että Ehlers-Danlos oireyhtymällä pitäisi olla jokaisella potilaalla seurantaohjelma nimenomaan sydämen ja verisuoniston osalta. Miten usein se tehdään ja miten se tehdään, niin se riippuu paljon tuosta sydänlääkäristä ja siitä mitä löydetään kun tutkitaan. Jos sydän on normaali eikä aortassa ole mitään viitettä, niin silloin esim. viiden (5) vuoden välein tapahtuva seuranta on ihan hyvä. Jos sieltä löytyy jotain, niin silloin siitä löydöksestä riippuen täytyy tehdä tutkimuksia useammin, koska moniin näihin ongelmiin on olemassa hoito. Jos aortta lähtee laajenemaan, niin on jopa mahdollisuus tehdä aorttaproteesi. Jos läppä alkaa vuotaa, niin voidaan laittaa keinoläppä. Jos on rytmihäiriöitä, niin niitäkin on Ehlers-Danlos oireyhtymässä, voidaan laittaa esim. tahdistin.
Kysymys: Missä iässä sydän- ja verisuoniongelmia yleensä esiintyy Ehlers-Danlosin syndroomassa?
Ei ole mitään sääntöä olemassa.
Kun Ehlers-Danlos diagnoosi asetetaan
Minusta silloin, kun Ehlers-Danlosin diagnoosi asetetaan, niin silloin täytyy tehdä tietty perustakartoitus. Tutkitaan luustoa ja ruuansulatuskanavaa tietyin tavoin, virtsaelimet ja tehdään sydämen- ja verisuonten ultraääni, sydänfilmitutkimus ja mahdollisesti magneettitutkimus.
Kysymys: Minkä alueen magneettitutkimuksesta on kysymys?
Silloin kun sydän tutkitaan, niin silloin pyydetään sydämen- ja verisuonten magneettitutkimusta.
Kommentti: Magneettitutkimukset ovat niin arvokkaita, ettei sinne saa lähetettä.
Tässä on juuri se lääkäreiden tietämys mistä on kysymys. Jos on vähäoireinen Ehlers-Danlos oireyhtymää sairastava nuori ihminen, niin sellainen tietty perustutkimus johon kuitenkin kuuluu sydämen ultraäänitutkimus pitää tehdä. Jos siellä havaitaan jotain poikkeavaa, niin silloin pitää edetä magneettitutkimukseen.
Kysymys: Sydänfilmissä epäillään infarktia. Samanlaisen käyrän aiheuttaa myös urheilutausta. Voiko Ehlers-Danlosin syndrooma aiheuttaa tällaisen samanlaisen koukeron sydänfilmiin?
Ei oikein voi. On mahdollista, että Ehlers-Danlos oireyhtymää sairastavalle vanhemmalla iällä ja erityisesti miehelle voi tulla sepelvaltimotauti. Se näkyy pienenä infarktimuotona. Jos on pelkästään sepelvaltimotauti ilman että se on aiheuttanut sydänlihakseen vaurioita, niin sydänfilmi on tavallisesti normaali. Lihasvaurio taas näkyy sydänfilmissä.
Kysymys: Kysyisin pään magneettikuvauksesta. Äidillä on ollut myös Ehlers-Danlos. Hänelle tuli suonenpullistuma ja aivoverenvuoto. Itselläni oli juuri pään magneettikuvaus. Vastauksessa aivotoiminta normaalia ja suonet normaalit. Lääkäri sanoi, että ei ole jatkotoimenpiteitä eikä seurantaa. Olisiko seuranta aiheellinen viiden vuoden välein?
Minusta se kuulostaisi oikein hyvältä. Jos kaikki nyt on kunnossa, niin silloin ei kannata kahden tai neljän vuoden kuluttua vaan juuri viiden vuoden kuluttua.
Kysymys: Minulla on sisäisiä verenvuotoja jatkuvasti. Juuri äskettäin oli tähystys peräsuolen kautta, ultraääni ja totesivat, että: “ei se kauhean pahalta näytä, hyvältähän se näyttää. Ei me oikeastaan löydetä mitään. Todennäköisesti ne pienen pienet verisuonet repeilee. Koeta pärjätä ja ota jotain vatsalääkettä”. Onko tämä ihan normaalia? Peijas-Rekolan sairaalan diagnoosi kesäkuun alusta (2008).
Tämä ei kuulosta yhtään turvalliselta eikö totta. On kai pakko hyväksyä se, että nyt tässä tilanteessa ei ole mitään sellaista aktiivia mitä voitaisiin tehdä. Olisi kuitenkin hyvä, että olisi vuoden tai kahden kuluttua joko uusi tutkimus tai ainakin suolistolääkärin uusi arvio mikä se tilanne on tällä hetkellä. Onko verenvuoto lisääntynyt, onko tullut lisäoireita, onko tullut devertikkeleitä eli pullistumia, jotka voisivat olla riski että suoli voisi jopa puhjeta.
Kysymys: Minkälainen poikkeavuus sydänfilmissä on tavallisin?
En osaa sanoa.
Kysymys jtk: Eli se ei ole mitenkään vaarallinen?
Mitä todennäköisimmin sydämen läpän vuotaessa esim. aorttaläpän vuotaessa se johtaa siihen, että sydänlihas paksunee ja sydämen asento muuttuu, niin silloin jo sydänfilmi muuttuu.
Kysymys jtk: Näkyykö siinä filmissä?
Kyllä se näkyy sydänfilmissä.
On muitakin harvinaisia, joita on ehkä kuvattu vain yksi tai kaksi potilasta maailmassa.
Nykyisin lääketieteellisiä julkaisuita on paljon, jotka ovat jakautuneet eri lääketieteen erikoisaloille. Nämä lehdet eivät nykyisin halua julkaista enää potilastapauksia, koska on päädytty siihen että niistä ei kuitenkaan opita yleissääntöjä. Kukaan ei ennätä ja jaksa lukea tapausselostuksia. Pitäisi olla joukko potilaita, jotka olisi tutkittu samoilla menetelmillä kontrolloidusti, asiantuntevasti, kirjoitettu hyvin ja selkeästi josta löytyy sellaista tietoa joka on laajemmalti hyödyksi niin potilailla kuin lääkäreille.
Ehlers-Danlosin oireyhtymä on harvinainen. Ei tulla ihan ajatelleeksi mitä kaikkea se merkitsee, vaikka me olemme koko ajan sivunneet näitä asioita. Tällä hetkellä tunnetaan noin 5000 – 6000 sairautta, jotka ovat harvinaisia yksittäisiä sairauksia. Mistä löytyy se tieto ja kokemus? Monissa näissä harvinaisissakin sairauksissa saattaa löytyä lääketieteellisiä julkaisuja, joista saadaan tietoa. Nykyisin on käytettävissä erinomaiset tietokannat, Internet mitä kautta se löytyy. Yksittäisellä lääkärillä yhden potilaan tavattuaan on monesti vaikeuksia päätellä että mitä minun pitäisi etsiä. Millä oireilla, millä nimellä mutta myös hänellä ei taida olla siihen aikaa. Tämäkin korostaa sitä, että tarvittaisiin keskuksia, jotka spesialisoituvat johonkin yhteen asiaan tai joukkoon asioita.
Tulee myös ongelma, että mistä löytyisi aika potilaan tai perheen hoitamiseen. Terveyskeskussysteemi on ajettu sellaiseen tilanteeseen, että useat lääkärit eivät halua tehdä siellä töitä. Johtuu siitä, että he kokevat että he eivät voi tehdä sitä työtä mikä heidän pitäisi siellä tehdä. Heillä on 15 – 20 min. aikaa potilasta kohden.
Meidän terveydenhuoltomme ongelmaa korostaa se, että yliopistolliset sairaalat ja keskussairaalat tekevät diagnooseja, antavat vähän ohjeita ja antavat jopa hyvätkin ohjeet ja lähettävät potilaan terveyskeskukseen, jossa lääkäri koettaa lukea potilaan papereista, että mitä kaikkea hänen pitäisi tehdä. Hän saa pitkän listan erilaisia asioita ja hänellä ei ole aikaa toteuttaa niitä kaikkia tai sitten ei ole niitä fasiliteetteja, sitä kuntoutusosastoa tai aikoja siellä vaikka hän haluaisi tämän tehdäkin.
Kysymys: Voiko terveyskeskuslääkäri lähettää perinnöllisyyslääketieteen yksikköön kuten HYKS:iin.
Voi lähettää.
Moniin pitkäaikaisiin sairauksiin liittyy vielä erityisongelmia, jotka tulevat vasta myöhemmässä vaiheessa. Kun harvinaisesta sairaudesta on kysymys, niin nimenomaan kun ei perusdiagnoosiakaan usein saada selville, niin ei osata arvioida ja arvata mitä kaikkea on viiden vuoden kuluttua tulossa. Puhumme silmistä, sydämestä, virtsarakosta, tällaisista asioista joissa ei nuorella iällä tarvitse olla mitään ongelmia, mutta jos tuntee sairauden niin tiettä, että tällaisia ongelmia voi tulla.
Harvinaisissa sairauksissa on myös se, että ei ole hoitokeinoja. Ei ole lääkettä. Ei ole koeteltuja, tutkittuja hoitomenetelmiä, mitkä asiat auttavat ja mistä on itse asiassa enemmän haittaa. Monesti tehdään laajasti erilaisia tutkimuksia, jotka monta kertaa “turhia”, joita ei kannattaisi tehdä, koska ne eivät tule kertomaan mitään tästä sairaudesta tai potilaasta. Kulutetaan paljon rahaa ja ollaan tutkivinaan. Vaikka yliopistollisessa sairaalassa saisikin asiantuntevan neuvonnan ja ohjeen, niin tiedon pitäisi vielä mennä kotipaikkakunnalle ja sitä toteutettaisiin, niin tässä ketjussa on usein katkoja.
Harvinainen sairaus – terveydenhuollon ongelmia
· Tiedon ja kokemuksen puute –
o Lähteitä on – mutta mistä aikaa/tukea tiedon hankkimiseen?
· Mistä aikaa potilaan/perheen hoitamiseen
· Miten huomioida sairauskohtaiset erityisongelmat
· Hoitokeinojen rajallisuus, näytön puute
· Hoitoketjujen puute/katkot
· Terveyden- ja sosiaalihuollon rajoitukset
Sosiaalihuollossamme kuten Kelassa tehdään erinomaista työtä, mutta varmaan melkein kaikilla Ehlers-Danlosin syndroomaa sairastavilla on kokemuksia siitä toivottomasta tilanteesta Kelan toimistossa, miten saada virkailijan ymmärtämään mistä tässä on kysymys, mitä tässä tarvitaan ja mikä olisi järkevää. Kelasta saa hakea tukea, mutta sen saaminen onkin jo vaikeampaa. Saa tapella erilaisten tukimuotojen saamiseksi, jotka kuitenkin meidän yhteiskunnassamme on aika hyvin järjestetty. On tietysti omat rajansa niin Kelassa kuin kunnankin Sosiaalitoimistossa, mutta siellä ei ole rahaa. Ei ole osattu arvioida, että itse asiassa tämä vaatii kustannuksia, henkilökuntaa ja kaikkea tätä. Tämä on harvinaisten sairauksien yksi ongelma.
Varsinkin perinnöllisyyslääkärille on tärkeää, että vaikka sairaus olisi kuinka harvinainen, niin se pitää pyrkiä diagnosoimaan tarkasti ja oikein.

Ehlers-Danlos oireyhtymän periytyvyys
Ehlers-Danlos oireyhtymä on periytyvä sairaus. Tunnetaan 5000 – 6000 erilaista perinnöllistä sairautta. Yli 2000 tunnetaan tarkasti, missä perintötekijävirhe on. On noin parituhatta sairautta, joissa tiedetään se paikka, mutta ei tiedetä missä se virhe oikeastaan on. On noin parituhatta sairautta, joissa ei ole aavistustakaan missä perintötekijävirhe on, mutta sukutiedot kertovat, että tämä on perinnöllinen sairaus eri tavoin periytyvänä. Nämä sairaudet ovat yhden perintötekijän mutaatioista johtuvia sairauksia. Tällä hetkellä ihmisellä on perintötekijöitä n. 20 000 – 25 000, mutta kukaan ei tarkalleen tiedä sitä. Kymmenen vuotta sitten kuvittelimme, että ihmisellä on 100 000 perintötekijää eli se on vähentynyt. Imettäväiset, hyönteiset, ihmiset perintötekijöiden suhteen eroaa yllättävän vähän toisistaan. Jotain ratkaisevaa siellä tietysti täytyy olla. Yhdessä tällaisessa perintötekijässä voi olla erilaisia mutaatioita ja se voi aiheuttaa erilaisia sairauksia tai vaikeusasteeltaan erilaisen saman sairauden. Periaatteessa Ehlers-Danlos oireyhtymät ovat kaikki perinnöllisiä, joiden perintötekijä mutaatiosta johtuvia. Ne jaetaan vallitsevasti periytyviin, peittävästi periytyviin ja on joukko sellaisia, jotka periytyvät sukupuoleen sidotusti. Olen sanonut, että Ehlers-Danlos oireyhtymän tyyppi V ei ole olemassa siinä luokittelussa, niin kyllä se tietyllä tavalla siellä on. Se aina pulpahtaa esiin osittain siksi, että se periytyy juuri sukupuoleen sidotusti. Tässä tuntuu olevan pientä ristiriitaa, mutta siitä ei pidä välittää. Se nyt on siellä. Tämä luokittelu on niin kuin oman periytyvyytensä kannalta ja nämä sairaudet ovat omaa luokkaansa.
Miten periytyvyys ja uusiutumisriski suvussa saadaan selville? Sukutiedot ovat erittäin tärkeitä. Jos me ajattelemme omia sukulaisiamme ja heidän mahdollisia sairauksia vähänkin syvemmältä niin huomaamme, että me emme tiedä sukulaisistamme juuri mitään. Tiedämmekö mihin isoisä tai isoäiti menehtyivät tai jommankumman veli menehtyi. Minkälaisia sairauksia heillä on ollut. Jos perinnöllisyyslääketieteen yksikköön joku on ohjattu ja ryhdytään tekemään sukuselvitystä, niin ei ole muuta keinoa kuin että me kysymme. Saamme tehtäväksi meidän asiakkaallemme tai potilaallemme koettaa selvittää sukulaistensa tilannetta. Tiedämme että se ole helppoa. Ei ole helppoa mennä kysymään sukulaisiltaan minkälaisia sairauksia sinulla on. Saatamme saada epäluotettavaa tai epätäydellistä tietoa. Kun kysymykset osataan asettaa oikein eli onko tätä ja tätä oiretta, niin alkaa muodostua sukupuu. Jos suvussa on sellaisia henkilöitä, joilla on sama sairaus.
Kysymykset saattavat joskus tuntua vähän epäasiallisilta kun kysytään, että onko verenvuotoa. Itse asiassa jos Ehlers-Danlos oireyhtymää ajatellaan, niin ei se ole epäasiallista. Kysymykset saattavat olla yllättäviä. Kun sukutietoa kyselemään, niin se on myös aikakysymys. Siihen menee tavattomasti aikaa, piirretään sukupuuta, kysytään hyvin täsmällisesti tiettyjen sääntöjen mukaan sukulaisten tietoja ja useimmiten täytyy vielä palata siihen, että potilas ottaa selvää ja tulee uudestaan.
Sukutiedoista voidaan päätellä monenlaista ja miten se periytyminen kaiken kaikkiaan menee. Jos periytymistapa sopii tähän epäiltyyn sairauteen, niin se on yksi tärkeä asia huomioida ja tietysti sairauden oireisto. Oireet ovat tärkeät. Tiedämme julkaisuista, että tähän sairauteen kuuluu tietyt oireet ja miten se tiettyjen sääntöjen mukaan.
Ihmisen perimä
· Jokaisessa ihmisen solussa on n. 25 000 perintötekijää eli geeniä
· Geenit on pakattu 46 kromosomiin, oikeastaan 23 kromosomipariin
· Geenien paikat kromosomeissa tunnetaan, yli 20 000 paikkaa tarkasti
· Geenien tehtävät tunnetaan vielä huonosti
Kysymys: Jos henkilöllä on todettu määrätyn tyyppinen Ehlers-Danlosin syndrooma. Hänellä on kolme lasta, joilla ei ole EDS:n oireita, niin voiko heidän lapsille tulla EDS?
On perhe jossa on kolme jälkeläistä ja näistä yhdellä on sairaus. Kysytään, että onko tämä perinnöllinen sairaus? Jos on vain tämä tieto, niin ei tästä voi sanoa mitään. Tämä sairaus voi kuitenkin olla perinnöllinen.
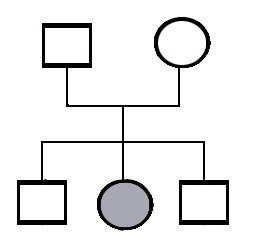
Periytyminen ?
Vallitseva periytyminen
Tässä on sama tilanne, mutta äidillä on tämä sama sairaus. Tässä tämä periytymistapa on jotakuinkin selvä. Tämä on vallitsevasti periytyvä sairaus. Ei tarvitse mennä taaksepäin sukupolvissa. Tämäntyyppinen periytymistapa jota kutsutaan vallitsevaksi periytymistavaksi. Sairaus voi tulla monen sukupolven läpi ja ilmenee suurin piirtein samalla tavalla ja silloin tiedämme, että se on vallitsevasti periytyvä.
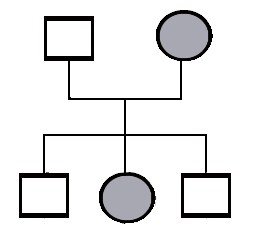
Vallitseva eli dominantti periytyminen

Peittyvä periytyminen
Tässä on toinen tilanne. Tässä taas vanhemmat ovat terveitä, mutta perheessä on kolme lasta ja kahdella on sama sairaus. Tämä jo viittaa siihen, että tässä voi olla kysymys peittyvästä periytymisestä. Tässä sairaudessa mitä todennäköisimmin vanhemmat ovat kaksi tervettä perintötekijän kantajaa. Heillä kahdesta mahdollisesta perintötekijästä - ihmisellä on kaikkia perintötekijöitä kaksi – toisessa on se vika. Kun se on vain toisessa, niin ihminen on terve mutta hän kantaa tuota perintötekijävirhettä. Jos kaksi tällaista ihmistä tapaa toisensa ja saa jälkeläisiä, niin on 25% todennäköisyys että he antavat molemmat samanaikaisesti sen perintötekijävirheen jälkeläiselleen. Silloin voi juuri tulla tämä tilanne että vanhemmat terveitä, mutta kolmesta jälkeläisestä esim. kaksi on sairasta.
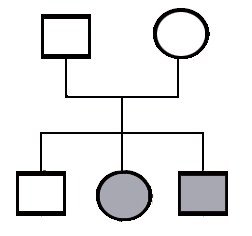
Peittyvä eli resessiivinen periytyminen

Kysymys: Voiko olla että vanhemmat on terveitä, mutta heidän vanhemmillaan on Ehlers-Danlosin sairaus?
Se on epätodennäköistä. Todennäköisempää on, että jommallakummalla se jolla tämä sama sairaus nyt on, on sairaus niin lievänä että hänellä sitä ei ole todettu. Se on yksi mahdollisuus.
Sukupuoleen sidottu periytymistapa kuten hemofilian (verenvuototauti) merkitsee sitä, että miehet sairastavat, naiset kantavat mutta ovat terveitä. Siinä tapahtuu sukupolven ylihyppääminen. Tuo sama perintötekijä voi siirtyä naiselta seuraavan polven naiselle ja seuraavankin polven naiselle, mutta jos hän saa lapsen joka on poika, niin tällä pojalla on 50% mahdollisuus saada tuon virheellisen perintötekijän. Silloin tässä suvussa voi siellä kauempana olla joku toinen mies jolla on ihan sama sairaus. Siinä hypätään yli.
On myös sellainen mahdollisuus, että jos on vallitsevasti periytyvä sairaus eli kuva jossa oli vain yksi sairas jälkeläinen, niin tällä yhdellä jälkeläisellä on hänen kohdallaan tapahtunut uusi mutaatio. Se aiheuttaa hänelle sairauden. Silloin vallitsevasti periytyvä sairaus ei ole kummallakaan vanhemmista eikä kenelläkään suvussa vaikka taaksepäin mentäisiin. Tällä yhdellä on sitten 50% riski saada sairas lapsi ja siitä lähtee tuo sukupolvesta seuraavaan meneminen.
Nämä ovat yksinkertaisia Mendelin sääntöjä, jotka voidaan perheelle tai potilaalle kertoa silloin kun tuo sairaus ja sukuselvitys on kunnolla tehty.

Tässä koeputkessa tuo pieni hahtuva on ihmisen perintötekijät. Siinä on DNA. Se on noin 10 ml verta erotetuista valkosoluista eristetty DNA. Siinä on 25 000 perintötekijää. Se on monesta solusta, niin niitä on moninkertaisesti sitä samaa perintötekijää. Perintötekijät on pakattu ihmisen kromosomeihin. Ihmisen jokaisessa solussa on 46 kromosomia. Oikeastaan pitäisi sanoa 23 paria sillä kaikkia kromosomipareja on siellä kaksi kappaletta. Samalla tavalla kaikkia perintötekijöitä on kaksi kappaletta. Nyt tällä hetkellä näistä 25 000 perintötekijästä noin 20 000 perintötekijää tunnetaan missä se on, mutta se tunnetaan vain kromosomin tarkkuudella. Kun on 23 kromosomia, niin tiedetään että se on tuossa kromosomissa, mutta jokaisessa kromosomissa on muutama tuhat perintötekijää. Ei tiedetä tarkalleen missä se siellä loppujen lopuksi on. Tätä työtä tehdään ja hyvää vauhtia mennään eteenpäin. Ongelmana on, että monesti perintötekijän tehtävä alkaa selvitä siitä, että siinä on joku vika. Se aiheuttaa jonkun sairauden tai ehkä jonkun ominaisuuden jolloin alkaa tuntua että tällä onkin joku tällainen tehtävä. Melkein jokaisella perintötekijällä on montakin erilaista tehtävää, mutta tästä ns. normaalista tehtävästä me tiedämme nykyisin vielä hyvin vähän. Tässä on molekyyligenetiikan ja perinnöllisyyslääketieteen suuri tehtävä ja tällaista tutkimusta tehdään aika hyvällä vauhdilla.
Jatkettu viimeksi 4.1.2009! Kirjoitus on vielä kesken!
Luento on nauhoitettu Reumaliiton kuntoutumiskeskuksessa Apilassa vuonna 2008. Tekstiä on hieman lyhennetty. Ehlers-Danlosin oireyhtymä on usein muutettu Ehlers-Danlosin syndroomaksi hakukoneissa paremman näkyvyyden vuoksi.
Kommentit